hpc:slurm
Table of Contents
Table of Contents
Slurm and job management
When we talk about cpu, we mean core of a cpu since all modern cpu are multi cores. So, one cpu = one core.
What is Slurm ?
According to Slurm official website :
Slurm is an open source, fault-tolerant, and highly scalable cluster management and job scheduling system for large and small Linux clusters. Slurm has three key functions.
- it allocates exclusive and/or non-exclusive access to resources (compute nodes) to users for some duration of time so they can perform work.
- it provides a framework for starting, executing, and monitoring work (normally a parallel job) on the set of allocated nodes.
- it arbitrates contention for resources by managing a queue of pending work.
Great, so what does it mean for the users of our HPC clusters ?
It means Slurm is the only way to being granted HPC resources. In order to request the resources you need to submit a job to Slurm or to ask it to allocate resources.
It is important to learn a few terms that are specific to Slurm :
- a resource: this may be cpu, gpu, running time (called wall time), memory, license or disk
- a job once resources are granted to you, your code is running on them and releases them when it finish. When you “submit a job”, you'll ask resources and your job will be put on waiting queue.
- a partitions determines on which resources your job will run
- a priority determine when you'll get access to a resource and your job will start
Resources
Before explaining more about Slurm jobs, it is important to understand to concept of partition and some other limits.
Partitions
What is a partition ?
A partition is a group of compute nodes accessible with some specific properties (such as maximum execution time, etc.).
- There are two main categories of partitions :
- public partitions are available for everybody on the clusters,
- private partitions are only available to their owners.
- When the HPC team buys compute nodes with UNIGE's funds, those nodes are 100% public which means they will be inside public partitions (
public-*).- Public partitions allow a maximum execution time of 4 days
- Public-short-* allows a maximum execution time of 1 hour, with a maximum of 6 cpu per jobs and 1 job running per user (multiple submit is allowed).
- When a research group buys compute nodes with their funds (private, FNS, etc.), those nodes will be inside both shared and private partitions.
- Private partitions give a higher priority to jobs (shorter waiting time) ;
- Private partitions also allow a maximum execution time of 7 days
- When the owner of the private partition is not using it, the nodes can be used by anyone else in the cluster since the nodes are shared in the shared partitions (
shared-*).- Hence, the (
shared-*) partitions contain all the cluster's nodes (public and private nodes) - Shared partitions allow a maximum execution time of 12 hours
Our clusters Baobab and Yggdrasil are heterogeneous, which means the nodes in a partition are not necessarily identical.
You can get a complete list of the nodes and their specifications in For advanced users - Compute nodes
Which partition for my job ?
This is a very important question, and choosing the right partition can save you (and others) a lot of time.
- Shared partions :
shared-cpu- for CPU jobs that need to run up to 12hshared-gpu- for GPU jobs that need to run up to 12hshared-bigmem- same asshared-cpu, but when you need a crazy amount of RAM (max. 12h )- N.B.
shared-*partition contain more nodes thanpublic-*(the sum of public and private nodes), but the maximum wall time is 12h.
- Public partitions:
public-cpu- for CPU jobs that need to run between 12h and 4 days (max. 4 days)public-gpu- for GPU jobs that need to run between 12h and 4 days (max. 4 days)public-bigmem- same aspublic-cpu, but when you need a crazy amount of RAM (max. 4 days)- N.B.
public-*partition contain less nodes thanshared-*, but the maximum wall time is 4 days.
- Special public partitions:
debug-cpu- to test your CPU jobs and make sure everything works fine (max. 15 min)public-interactive-gpu- Run interactive jobs or to test your GPU jobs and make sure everything works fine (max. 04h)public-interactive-cpu- for interactive CPU jobs (max. of 6 cores for 8h)public-longrun-cpu- for CPU jobs that don't need much resources, but need a longer runtime time (max. of 2 cores for 14 days)public-short-cpufor CPU jobs that need to run 1h max and 6 CPU max (limited to 1 job running per user but multiple submits allowed)
- Private partitions:
- The number of CPU/GPU nodes in a private partition depends on the partition
- If none of your colleagues are using the private partition, max. waiting time is 12h
- For jobs that need to run for more than 4 days (max. 7 days)
You can read more about job priorities and waiting time in the Slurm Job priority section.
Partitions lists
The command sinfo shows you more details about the partitions you have access to (default time, etc.)
[root@admin1 ~]# sinfo PARTITION AVAIL TIMELIMIT NODES STATE NODELIST debug-cpu* up 15:00 2 idle cpu[001-002] debug-gpu up 15:00 1 idle gpu001 public-cpu up 4-00:00:00 77 alloc cpu[006-082] public-bigmem up 4-00:00:00 4 idle cpu[112-115] public-gpu up 2-00:00:00 6 idle gpu[002-006,008] shared-cpu up 12:00:00 33 mix cpu[005,084-111,116-119] shared-cpu up 12:00:00 77 alloc cpu[006-082] [...]
The default partition is
debug-cpu (see, there is as '*' in its name). If you want to use another one, you must explicitly specify it.
In the list of partitions (see below), the “Max mem per core” is the suggested value, but it is not enforced. Please refer to the Memory and CPU usage section for details about how to request a specific amount of memory. If you need more than 10GB RAM per core, you might want to use one of the *-bigmem partition.
Clusters partitions
As of 2020-11, the new partition naming is available on Baobab.
| Partition | Time Limit | Max mem per core |
|---|---|---|
| debug-cpu | 15 Minutes | full node memory |
| public-interactive-gpu | 4 hours | full node memory |
| public-interactive-cpu | 8 hours | 10GB |
| public-longrun-cpu | 14 Days | 10GB |
| public-cpu | 4 Days | 10GB |
| public-gpu | 4 Days | 10GB |
| public-bigmem | 4 Days | half node memory |
| shared-cpu | 12 Hours | 10GB |
| shared-gpu | 12 Hours | 10GB |
| shared-bigmem | 12 Hours | 500GB |
All the compute nodes have 3GB of memory per core by default. Minimum resource is one core.
N.B. : no public-interactive-gpu, nor public-gpu partitions on Baobab, as there are only private GPU nodes.
Private partitions
To avoid confusion, private partitions aren't detailed here.
| Partition | Time Limit | Max mem per core | default Mem Per core |
|---|---|---|---|
| private-<privatename> | 7 Days | full node memory | 3GB |
Wall time
When you submit a job, you need to specify the estimated duration of your job.
The time formats accepted by Slurm are as follows:
minutes minutes:seconds hours:minutes:seconds days-hours days-hours:minutes days-hours:minutes:seconds
Example:
1-12:30:25
Means: 1 day, 12 hour, 30 min and 25 seconds
Make sure you estimate the amount of time you correctly for your job and make sure the partition you use allows to run your code for this duration. Your job has more chance to start quickly if you ask less resources.
Memory
When you submit a job, the usable memory you have is 3GB per core. If you are running a job which requires more or less memory per core, you can specify it like this:
--mem-per-cpu=1000 # in MB
Even if you have requested a full node, you still need to specify how much memory you need:
--mem=60000 # 60'000 MB => 60 GB
This is even the case if you request a partition such as *-bigmem!
The value of 0 will request all the node's memory.
You can use
--mem=0 to ensure you use the entire memory of a node.
GPU
Currently on Baobab and Yggdrasil there are several nodes equipped with GPUs. To request a GPU, it's not enough to specify a partition with nodes having GPUs, you must as well specify how many GPUs and optionally the needed GPU type.
To specify how many GPU to request, use the option --gpus=n with n having a value between 1 and the maximum according to the table below.
You should also specify the type of GPU you want:
- ampere, high end architecture for multipurpose use
- titan, for single precision computation, like machine learning
- pascal, for double precision computation like physical simulations
- rtx, to accelerate machine learning, data science workflows and ray tracing
Example to request three titan cards: --gpus=titan:3.
You can find a detailed list of GPUs available on our clusters here : https://doc.eresearch.unige.ch/hpc/hpc_clusters#for_advanced_users
Resources :
CPU
You can request all the CPUs of a compute node minus two that are reserved for the OS. See slurm core spec
CPU types
It's normally not important on which type of node your job is running. But there are some cases where it is important to be able to stick to a given kind of CPU or a certain generation of CPUs.
You can request for example to have only nodes with CPU E5-2660V0 :
srun --constraint=E5-2660V0 or #SBATCH --constraint=E5-2660V0
Or you can specify that you want a node of generation V3 :
srun --constraint=V3 or #SBATCH --constraint=V3
You can specify as well multiple constraints using logical or with the symbol |. For example if you don't want to use nodes of generation V1:
srun --constraint="V2|V3|V4|V5|V6" or #SBATCH --constraint="V2|V3|V4|V5|V6"
If you want a list of those specifications, please check : For advanced users - Compute nodes
Single thread vs multi thread vs distributed jobs
There are three job categories each with different needs:
| Job type | Number of cpu used | Examples | Keywords | Slurm |
|---|---|---|---|---|
| single threaded | one CPU | Python, plain R | - | |
| multi threaded | all the CPUs of a compute node (best case scenario) | Matlab, Stata-MP | OpenMP, SMP | --cpus-per-tasks |
| distributed | can spread tasks on multiple compute nodes | Palabos OpenFOAM | OpenMPI, workers | --ntasks |
There are also hybrid jobs, where each tasks of such a job behave like a multi-threaded job. This is not very common and we won't cover this case.
In slurm, you have two options for asking CPU resources:
--cpus-per-tasks: this will specify that you want more than one CPU per task.--ntasks: this will launch n time your job. ONLY specify a value bigger than one if your job knows how to handle multitasking properly. For example OpenMPI job can benefit of this option. If your job doesn't handle this option correctly, it will be launched n time doing strictly the same things, this is not what you want and will wait resources and create corrupted output files.
Submitting jobs
After connecting to the login node, you need to submit a job to Slurm to request computing resources. You can request it in 3 ways :
| method | execute job | blocking command | support job array | batch script needed |
|---|---|---|---|---|
| salloc | no | yes | no | no |
| srun | yes, single one | yes | no | no |
| sbatch | yes | no | yes | yes (or -w option) |
By blocking command, we mean that you need to keep your terminal open during the whole process. If you close it, your job will finish.
An “sbatch” script is a Bash script with special instructions for Slurm.
Do not execute directly the sbatch script as bash script, but submit it using
sbatch (otherwise your script will be launched on the login node and this is forbidden):
[sagon@login2 slurm] $ sbatch example.sh Submitted batch job 40721231
The number 40721231 is the jobid that is a uniq number. You can use it later for reporting, see logs or cancel for example.
Whatever method you use, you need to pass some arguments (such as how many CPU you need, which Slurm partition to use, how long your script will run, how to execute your code). Slurm will then add your job in a queue with other users' jobs, and find the fastest way to provide the resources you asked for. When the resources are available, your job will start.
There are various possibilities to submit a job on the cluster. We have detailed the most common usage below.
If you belong to more than one account, you should specify which account should be used with the option
--account=<account>
Batch mode (sbatch)
To use the batch mode, you need a sbatch script. Here is an Example of an sbatch script my_sbatch.s with #SBATCH directives :
#!/bin/sh #SBATCH --job-name jobname # this is a parameter to help you sort your job when listing it #SBATCH --error jobname-error.e%j # optional. By default a file slurm-{jobid}.out will be created #SBATCH --output jobname-out.o%j # optional. By default the error and output files are merged #SBATCH --ntasks 1 # number of tasks in your job. One by default #SBATCH --cpus-per-task 1 # number of cpus for each task. One by default #SBATCH --partition debug-cpu # the partition to use. By default debug-cpu #SBATCH --time 15:00 # maximum run time. module load my_software # load a specific software using module, for example Python srun my_software # run your software
To submit your job proceed as follows:
[sagon@login1 ~]$ sbatch example.sh Submitted batch job 53697
The job number can be useful if you want to see the status of the job on the queue of kill it for example.
To have a list of all options, please check Slurm documentation - sbatch
You can submit a batch job either with the help of a script or directly by specifying the options on the command line. The recommended way to use options for sbatch is to add them directly in the script as in the previous example.
Here are some examples to illustrate the second possibilities.
A simple example to launch a batch job my_sbatch.sh using 16 cpus on the default partition:
sbatch --ntasks=16 my_sbatch.sh
If not specified, the default partition is
debug-cpu and the the default number of cpus per task is 1
Launch a job specifying the partition “shared-cpu” (see Partitions and limits section) and max execution time:
sbatch --ntasks=8 --partition=shared-cpu --time=0-00:05:00 my_sbatch.sh
You will have more chance to have your job quickly scheduled if you specify an accurate max execution time (i.e. not the permitted maximum).
Monothreaded jobs
Click here for job type comparison.
When you submit a job on the clusters, the minimum resources that are allocated to you is a one cpu. As the node may be shared with other users, it is wise to specify the amount of memory you need per core. If you don't, the default memory allocated is 3GB per core.
Let's say you want to launch one job that needs 1GB per core. Your sbatch script is named example.sh
#!/bin/bash #SBATCH --partition=public-cpu #SBATCH --time=05:00 #SBATCH --ntasks=1 #SBATCH --mem-per-cpu=1000 # in MB srun ./yourprog
To submit your job do as follows:
[sagon@login1 ~]$ sbatch example.sh Submitted batch job 53697
The number you get is the jobid.
Adapt this example to fit your needs. If you need to scale this solution to a bigger number of similar tasks, see the Job array section.
Multithreaded jobs
Click here for job type comparison.
When you have a program that needs more than one core per task (openMP, STATA etc), you can adapt the Monothreaded jobs example by adding one line:
#SBATCH --cpus-per-task=x
where x is between 1 and 128. We have only a couple of AMD nodes with 128 cores. If you request a high number of cpus per task, be sure that you need it as you may have to wait many days for them to be available.
If you want to use all the cores of a node, but you don't know in advance the characteristic of the node, you can use this sbatch script:
#!/bin/sh #SBATCH --job-name=test #SBATCH --time=00:15:00 #SBATCH --partition=public-cpu #SBATCH --output=slurm-%J.out #SBATCH --exclusive #SBATCH --ntasks=1 # We want to have a node with minimum # 2 sockets, 4 cores per socket and 1 thread per core # nb_cores = 2 * 4 * 1 = 8 cores # if you want more cores, you can increase the number of cores per socket to # 6 or 8 to have 12 or 16 cores. #SBATCH --extra-node-info=2:4:1 We want to have at least 12GB RAM on this node #SBATCH --mem=12000 # run one task which use all the cpus of the node srun --cpu_bind=mask_cpu:0xffffffff ./mySoft
Distributed jobs
When you want to distribute your jobs across nodes, your software will probably use an MPI implementation such as OpenMPI or Intel.
To specify the number of MPI workers, you need to specify the number of tasks in slurm.
--ntasks-per-node: maximum number of tasks per node. Can be used in conjunction with --ntasks.
-n, --ntasks=<number>: number of workers in total
-N, --nodes=<minnodes[-maxnodes]>: number of nodes allocated to this job
You can have as well a hybrid job requiring tasks and cpus (for OpenMPI + OpenMP for example).
If you request for example 20 tasks and 1 CPU per task, your job may be executed by one compute node having 20 cpus if available or may be spread across any number of compute nodes. The issue may be that the compute nodes are shared with other jobs and not from the same generation.
To circumvent this, you can specify the cpu type you want, but you'll have more wait time.
You may as well want to specify the --exclusive flag to be sure that your compute node won't be shared
with other jobs. In this case, please do not specify the number of compute node or you may waste resources.
You may as well be interested by the distribution parameter.
--distribution: Specify alternate distribution methods for remote processes.
If you need more details, please consult the sbatch man page.
If you want to compile your software against MPI, it is very important not to compile using directly gcc, icc or similar commands,
but rather rely on the wrappers mpicc, mpic++, mpicxx or similar ones provided by
module.
Remember that you need to load the same module at runtime as well.
See here for more information about the toolchains
GPGPU jobs
When we talk about GPGPU we mean using a GPU to perform calculation, not for visualization.
You can see on this table How our clusters work all the GPUs models we have on the cluster. You may notice that we have a very wide range of GPU models, from low end to high end. It is important to select the correct GPU model to avoid to waste resources. The important characteristics of a GPU are:
- on board memory in GB
- simple precision vs double precision for float calculation
- compute capability
Specify the memory needed. For example, request one GPU that has 10G at least.
srun --gres=gpu:1,VramPerGpu:10G
If you just need a GPU and you don't care of the type, don't specify it. You'll get the lower model available.
#SBATCH --gpus=1
Example to request two double precision GPU model:
#!/bin/sh #SBATCH --partition=shared-gpu #SBATCH --gpus=2 #SBATCH --constraint=DOUBLE_PRECISION_GPU srun nvidia-smi
It's not possible to put two types in the GRES request, but you can ask for specific compute capability, for example you want to request any GPU model with compute capability bigger or equal to 7.5:
Example
#!/bin/sh #SBATCH --partition=shared-gpu #SBATCH --gpus=1 #SBATCH --constraint="COMPUTE_CAPABILITY_7_5|COMPUTE_CAPABILITY_8_0|COMPUTE_CAPABILITY_8_6|"
Example of script (see also https://gitlab.unige.ch/hpc/softs/tree/master/c/cuda ):
#!/bin/env bash #SBATCH --partition=shared-gpu #SBATCH --time=01:00:00 #SBATCH --gpus=titan:1 module load CUDA # see here for more samples: # /opt/cudasample/NVIDIA_CUDA-8.0_Samples/bin/x86_64/linux/release/ # if you need to know the allocated CUDA device, you can obtain it here: echo $CUDA_VISIBLE_DEVICES srun deviceQuery
If you want to see what GPUs are in use in a given node:
scontrol -d show node gpu002 [...] Gres=gpu:titan:3 GresUsed=mic:0,gpu:titan:3(IDX:0-2) [...]
In this case, this means that node gpu002 has three Titan cards, and all of them are allocated.
Interactive jobs
If you need to perform some debug/tests or if your software requires a graphical interface,
you can start an interactive session on a compute node.
For example, let's say you want to start a session on the debug-cpu partition for a duration of
15 minutes, using 2 CPUs:
[sagon@login2 ~]$ salloc -n1 -c2 --partition=debug-cpu --time=15:00 --x11
When the resources are allocated to you, you can see the login prompt change to reflect that you are using a compute node and not the login node:
[sagon@nodexxx ~]$
When done, you can stop the session like this:
[sagon@nodexxx ~]$ exit
Adding the option
--x11 will allow you to submit graphical job
For interactive programs, you might want to use the
public-interactive-cpu partition (Which partition for my job)
Job array
Slurm supports natively the notion of job arrays. A job array is useful when you have a lot of the same jobs to launch and you just want to give a different parameter to every job.
Here is an example for launching n time a monothreaded job.
The following sbatch script my_job_array.sh is used and the variable ${SLURM_ARRAY_TASK_ID} shows the taks ID in the job array:
#!/bin/bash #SBATCH --partition=shared-cpu #SBATCH --time=00:10:00 #SBATCH --cpus-per-task=1 #SBATCH --ntasks=1 #SBATCH --mem-per-cpu=1000 # in MB #SBATCH -o myjob-%A_%a.out srun echo "I'm task_id " ${SLURM_ARRAY_TASK_ID} " on node " $(hostname)
When you want to execute a job array, use the --array option. You may specify how many cores ONE instance of your array needs with --ntasks if you are using MPI or with --cpus-per-task otherwise. You must specify the array size and offset by specifying the start and stop of the array index.
N.B. The maximum size of an array is currently set to 10 000.
We encourgae you to limit the maximum number of simultaneously running tasks from the job array may be specified using a “%” separator.
For example “- -array=0-15%4” will limit the number of simultaneously running tasks from this job array to 4
Example to launch an array of 100 jobs, each one using one core:
sbatch --array=1-100%4 my_job_array.sh
The max number %100 sems to be a good limit :)
Slurm Quota
Overview
Slurm Quota allows you to limit compute usage on the HPC cluster.
Quotas are set by the HPC team upon request.
Quota Levels
In the following example, the PI Sidious manages two users: Vador and Maul.
User Level
Sidious ├── Vador → ⛔ limited (quota reached) └── Maul → ✅ not affected
- Only one user (
Vador) is limited - Other users continue normally
Error message at submission:
srun: error: User quota exceeded srun: error: Unable to allocate resources: Access/permission denied
Account Level
Sidious (quota reached) ├── Vador → ⛔ blocked └── Maul → ⛔ blocked
- The quota is set on the Slurm account (
Sidious) - All users are impacted
- No job submission is allowed once the limit is reached
How It Works
Submit job
│
▼
Check quota
│
┌───┴──────────┐
▼ ▼
✅ OK ⛔ Exceeded
│ │
▼ ▼
Run job Reject job
│
▼
Usage updated (after job)
- Quota is checked at submission time
- Subsequent submissions will be blocked
⚠️ Note: The quota is checked at submission time. A job may exceed the quota if it was still below the limit when submitted. Once usage is updated and the quota is reached, all new submissions will be blocked.
Why Use It
- Control usage
- Avoid unexpected costs
- Protect project budgets
Request a Quota
Contact the HPC team with:
- Target (user or account)
- Wanted limit
- Quotas are initiated upon request from the PI
- They can later be modified:
- By the PI
- By the user (only to set a more restrictive limit)
- Account-level quotas can only be modified by the PI
Advanced usage
Job dependency
Slurm can handle job dependencies in various way.
Some of the dependency features available in Slurm (cf. man sbatch)
after:job_id[[+time][:jobid[+time]...]]afterany:job_id[:jobid...]afternotok:job_id[:jobid...]afterok:job_id[:jobid...]expand:job_idsingleton
Example to submit multiple batch jobs that are dependent from each other. The dependent job will only start when the previous one terminated with success:
[sagon@login2 ~]$ sbatch pre_process.bash Submitted batch job nnnnn [sagon@login2 ~]$ sbatch --dependency=afterok:nnnnn do_work.bash Submitted batch job 12346
N.B. in this example, the first submitted job is given the job ID “nnnnn”
(in real life, it will be a number), and the second one depends on it thanks to the option
--dependency=afterok:nnnnn. You obviously need to replace nnnnn with the job ID returned by Slurm.
You can see here an example with singleton used to restart a job when time limit is reached.
Master/Slave
You can run different job step in an allocation. This may be useful in case of a master/slave program for example.
For example, if you want to launch a master program on core 0 and 4 slaves jobs on cores 1-4:
[sagon@login2 ~]$ cat master.conf #TaskID Program Arguments 0 masterprogramm 1-4 slaveprogramm --rank=%0 [sagon@login2 ~]$ srun --ntasks=5 --multi-prog master.conf
Use %t and %o to obtain respectively the task id and offset.
Checkpoint
A checkpoint allows you to pause/stop a program and to restart it from the “saved” point. This is usually done by saving your progress to a file, and your program can be restarted from that file.
Not all programs implement checkpointing, so you have to check if this is an option for you.
If you can use checkpoints, this can be interesting :
- to save your progress and avoid starting all over again if your job fails.
- to run a time-consuming task in multiple short jobs to obtain resources faster
- instead of requesting a job for 4 days, you could execute it by running multiple 12h jobs consecutively. This could decrease your waiting time to obtain the resources (as it's usually faster to get a slot of 12h than it is for 4 days).
Please check this post :
Reservation
to request for reservation; contact the HPC team following the instruction https://doc.eresearch.unige.ch/hpc/start > contact the hpc team by email
list reservation:
(baobab)-[alberta@login2 ~]#scontrol show res
Use reservation via srun:
(baobab)-[alberta@login2 ~]# srun --reservation <reservation_name> hostname
Use reservation via script sbatch:
#SBATCH --reservation <reservation_name>
#!/bin/bash #SBATCH --job-name=test_unitaire #SBATCH --reservation test srun hostname
Job monitoring
If you want to get an estimated start time for your job, you can use --test-only :
srun --test-only -n100 -p public-cpu -t 10 hostname
To see the queue :
squeue
Add the -u <username> to see a list of all your job together with the node they are scheduled on and the reason why they might not be starting.
Add the –start flag to obtain the scheduled time of when your job should start. 1)
squeue -u $USER --start
To get an estimation of when your job is scheduled to start :
scontrol show jobid 5543
Those commands give an estimation. Sometimes the waiting time will be longer, sometimes it will be shorter
Email notification of job events
When a determined event occurs in your job's life, it is possible to receive an email to the address used during your Account registration :
--mail-type=<type> BEGIN, END, FAIL, REQUEUE, and ALL (any state change).
Those options can be used with sbatch and srun.
If your job get killed, one of the reason could be that you have used too much memory. To check if it's the case, you can have a look at dmesg.
For instance :
dmesg Memory cgroup out of memory: Kill process 62310 (cavity3d) score 69 or sacrifice child
The email you receive contains important information about why your job failed for instance, but it's not always easy to read it. For example in this excerpt, you need to look inside the big block at th beginning to find the Reason PartitionTimeLimit (meaning the requested time is too long for the requested partition) :
[...] $ scontrol show Job=123456 JobId=123456 JobName=/home/users/b/brero/testFailed.sh [...] JobState=PENDING Reason=PartitionTimeLimit Dependency=(null) Requeue=1 Restarts=0 BatchFlag=1 Reboot=0 ExitCode=0:0 RunTime=00:00:00 TimeLimit=5-13:20:00 TimeMin=N/A [...]
Memory and CPU usage
You can see how much memory/cpu your job is using if it is still running using sstat. :
sstat --format=AveCPU,MaxRSS,JobID,NodeList -j <yourjobid>
If your job is no longer running, you can use sacct to see stats about. :
sacct --format=Start,AveCPU,State,MaxRSS,JobID,NodeList,ReqMem --units=G -j <yourjobid>
If you want other information please see the sacct manpage.
by default the command displays a lot of fields. You can use this trick to display them correctly. Then you can move with left and right arrows to see the remaining fields
(yggdrasil)-[root@admin1 ~]$ sstat -j 39919765 --all | less -#2 -N -S
1 JobID MaxVMSize MaxVMSizeNode MaxVMSizeTask AveVMSize MaxRSS MaxRSSNode MaxRSSTask AveRSS MaxPages MaxPagesNode MaxPagesTask AvePages MinCPU MinCPUNode MinCPUTask AveCPU NTasks AveCPUFreq ReqCPUF>
2 ------------ ---------- -------------- -------------- ---------- ---------- ---------- ---------- ---------- -------- ------------ -------------- ---------- ---------- ---------- ---------- ---------- -------- ---------- ------->
3 39919765.ex+ 489808K cpu095 0 5584K 1728K cpu095 0 1728K 0 cpu095 0 0 00:00:00 cpu095 0 00:00:00 1 2.80M >
4 39919765.ba+ 1298188K cpu095 0 1298188K 599588K cpu095 0 599588K 2511 cpu095 0 2511 00:39:25 cpu095 0 00:39:25 1 984K >
Energy usage
CPUs
You can see the energy consumption of your jobs on Yggdrasil (Baobab soon). The energy is shown in Joules using sacct.
(yggdrasil)-[root@admin1 state] (master *)$ sacct --format=Start,State,JobID,ConsumedEnergy,ConsumedEnergyRaw --units=k -j 28478878
Start State JobID ConsumedEnergy ConsumedEnergyRaw
------------------- ---------- ------------ -------------- -----------------
2023-10-12T09:48:28 COMPLETED 28478878 43.13K 43127
2023-10-12T09:48:28 COMPLETED 28478878.ex+ 43.13K 43127
2023-10-12T09:48:28 COMPLETED 28478878.0 43.11K 43109
It is working only for Intel nodes (at least for the moment). Only in the case of an exclusive job allocation does this value reflect the job's real energy consumption.
GPUs
If you are interested by the power usage of a GPU card your job is using, you can issue the following command while your job is running on a GPU node:
(baobab)-[root@gpu002 ~]$ nvidia-smi dmon --select p --id 0
# gpu pwr gtemp mtemp
# Idx W C C
0 63 55 -
0 59 55 -
0 62 55 -
Other tools
spart
This tool isn't working anymore and it seems a dead project
spart 2) is a tool to check the overall partition usage/description.
More info in this post.
[brero@login2 ~]$ spart
QUEUE STA FREE TOTAL RESORC OTHER FREE TOTAL || MAX DEFAULT MAXIMUM CORES NODE
PARTITION TUS CORES CORES PENDNG PENDNG NODES NODES || NODES JOB-TIME JOB-TIME /NODE MEM-GB
debug-EL7 * 32 64 50114 1 2 4 || 2 15 mins 15 mins 16 64
mono-EL7 120 784 4197 43 0 49 || - 1 mins 4 days 16 64
parallel-EL7 120 784 2108 18 0 49 || - 1 mins 4 days 16 64
shared-EL7 326 3572 734 489 1 224 || - 1 mins 12 hour 12 40
mono-shared-EL7 326 3572 144348 615 1 224 || - 1 mins 12 hour 12 40
bigmem-EL7 14 16 66 0 0 1 || 1 1 mins 4 days 16 256
shared-bigmem-EL7 63 212 820 0 1 10 || - 1 mins 12 hour 8 256
shared-gpu-EL7 415 484 16 36 3 12 || - 1 mins 12 hour 12 128
admin-EL7 16 16 0 0 1 1 || - 1 mins 7 days 16 64
YOUR YOUR YOUR YOUR
RUN PEND OTHR TOTL
COMMON VALUES: 0 0 0 0
pestat
pestat 3) is another tool to check cluster usage, this time focusing on single nodes
More info in this post.
[brero@login2 ~]$ pestat
Hostname Partition Node Num_CPU CPUload Memsize Freemem Joblist
State Use/Tot (MB) (MB) JobId User ...
gpu002 shared-gpu-EL7+ mix 6 12 8.29* 257820 231499 39803837 krivachy 39806932 salamda0 39795512 blonde0 39795511 blonde0 39795510 blonde0 39795480 blonde0
gpu004 shared-gpu-EL7 mix 6 20 7.27* 128820 102173 39795515 blonde0 39795516 blonde0 39795485 blonde0 39795481 blonde0 39795482 blonde0 39795499 blonde0
gpu005 shared-gpu-EL7 mix 5 20 6.05* 128820 97620 39795513 blonde0 39795463 blonde0 39795464 blonde0
[...]
node001 debug-EL7* idle 0 16 8.64* 64000 47239
node002 debug-EL7* alloc 16 16 7.63* 64000 16393 39772071 gonzalcr
node003 debug-EL7* alloc 16 16 11.16* 64000 18610 39772072 gonzalcr
node004 debug-EL7* drain* 0 16 0.01 64000 62061
node005 mono-EL7 drng* 8 16 2.52* 64000 38015 39802885 vie
node007 parallel-EL7 alloc 16 16 16.03 64000 53761 39524658 robinsh8
node008 mono-EL7 mix 8 16 2.13* 64000 53388 39802883 vie
node009 parallel-EL7 alloc 16 16 0.01* 64000 54509 39524658 robinsh8
node010 parallel-EL7 alloc 16 16 0.01* 64000 49796 39524658 robinsh8
node011 mono-EL7 mix 8 16 1.53* 64000 48417 39802889 vie
seff
seff is tool get a Slurm job efficiency report
[sagon@login2 ~] $ seff 30455298 Job ID: 30455298 Cluster: baobab User/Group: savchenk/hpc_users State: COMPLETED (exit code 0) Cores: 1 CPU Utilized: 00:00:01 CPU Efficiency: 0.77% of 00:02:10 core-walltime Job Wall-clock time: 00:02:10 Memory Utilized: 34.16 MB Memory Efficiency: 1.14% of 2.93 GB
HDF5 profiling plugin
The HDF5 plugin can help profile your jobs 4).
Official documentation :
The HDF5 files will be saved as /opt/cluster/slurm/hdf5/${USERNAME}/${JOBID}.h5.
You can analyse the HDF5 files directly on the login node via HDFView 5).
Cancel jobs
If you realise you made a mistake in your code or in your sbatch script, you can cancel your pending jobs or your running job (only yours).
If you want to cancel a particular job, you need to know its job id (you can see it on the web interface or using squeue).
Cancelling a job using its job id:
scancel <jobid>
Cancel a job and send a custom signal (for example to force the binary to generate a checkpoint):
scancel --signal=signal_name
You can read this topic Gracefully quit when job is cancelled
Cancel all the jobs belonging to one user that are in a pending state:
scancel --user=meyerx --state=pending
Job priorities
How is the priority of a job determined ?
HPC users often wonder how is the job priority calculated by Slurm.
On our clusters, the priority is obtained in general by four criteria, and each criterion has a different weight.
partition : 15000
age: 300
jobsize: 1000
fairshare: 30000
- The
partitioncriterion is always the same for a given user on a given partition. It is higher if you use a private partition.- For example, if you submit a job to the partition
shared-cpuyou’ll get a priority of 3750 for thepartitioncriterion. - However, this
partitioncriterion may have a multiplier, ie : if you use a private partitions. - For example, if a user submit a job on a private partition, the multiplication factor is 4 and he will have a priority of 15000 for the
partitioncriterion. - The reason for this is because private nodes are simultaneously part of multiple partitions : a private partition and
shared-cpupartition. The multiplier ensures that the people with access to private partitions have a faster access to their private node.
- The
agecriterion starts to increase as soon as the job is submitted. Hence, the priority will slowly increase with time. This is used to prevent a job with a low priority to stay in the queue for ages. - The
jobsizecriterion is here to favour big jobs, but the weigh is very low unless you submit a very big job. - The
fairsharecriterion is based on the past usage of the user. If you use the cluster a lot, yourfairsharecriterion will decrease. Every 2 weeks, half of your past usage is “forgotten” by Slurm.
You can see how the priority of a job and see how it is calculated with the sprio command. Example
- for a public partition (partition weight 3750)
[root@master ~]# sprio -j xxx JOBID PARTITION PRIORITY SITE AGE FAIRSHARE JOBSIZE PARTITION QOS xxx mono-shar 3810 0 6 53 2 3750 0 - for a private partition (partition weight 15000)
[root@master ~]# sprio -j xxx JOBID PARTITION PRIORITY SITE AGE FAIRSHARE JOBSIZE PARTITION QOS yyy wesolowsk 19590 0 1 4586 4 15000 0 zzz dpnc-EL7 21288 0 0 6297 2 15000 0
The priority is calculated by summing the values of all four criteria (it's actually a little more complex than that, as the priority returned by sprio isn't an exact sum, but you get the general idea). The bigger the priority, the higher is your chance to be scheduled compared to a job with a lower priority. The partition and fairshare criteria have then the strongest impact on the total priority calculation.
If you want to learn more about it, you can check this presentation :
- Slurm Priority, Fairshare and Fair Tree (might differ from how we do things on UNIGE clusters)
Priority vs. waiting time
Please also remember that the priority of a job is one thing, but the actual waiting time is another.
- Your priority is calculated regardless of the amount of resources you request for a job.
- If you request a large amount of resources, it might take more time for Slurm to provide those resources (the time to wait until they are free).
- Hence, asking for more or less memory won't change the priority, but asking for more resources might affect the waiting time.
- So it is better to request as little resources as possible if you want increase your chances to be scheduled quickly.
The waiting time is a pessimistic estimation and is based on the maximum time a job is announced to take. If you don't use the #SBATCH –time directive in your sbatch script, the default time for a job is given by the maximum time limit of the partition (12h, 4 day or 7 days depending on the partition). This implies that a job with a maximum run time of 4 days will be scheduled to take the whole 4 days and other jobs will be scheduled to run after it has finish. However if the job only take 10 hours the other jobs can start sooner. 6)
However, if you are using a shared-* partition, bear in mind that a new job on a private partition might “cut in line” as they have a higher priority.
If you are requesting for example 12h00 of wall-time and 16 cores, and another user is asking for a wall-time of 1 minute and 1 core, this job will most likely be scheduled before your job thanks to the backfill mechanism even if you job has a higher priority.
The total number of running jobs of a given user doesn’t change the priority of a job. But if the user uses the cluster a lot (especially a lot in a short time), then his/her fairshare criterion will decrease.
Please also check this post : HPC Community - Job priority explanation
Backfill mechanism
The priority is determined by various factors like the usage history, job age and its size.
The scheduler use a backfill to maximize the cluster usage.
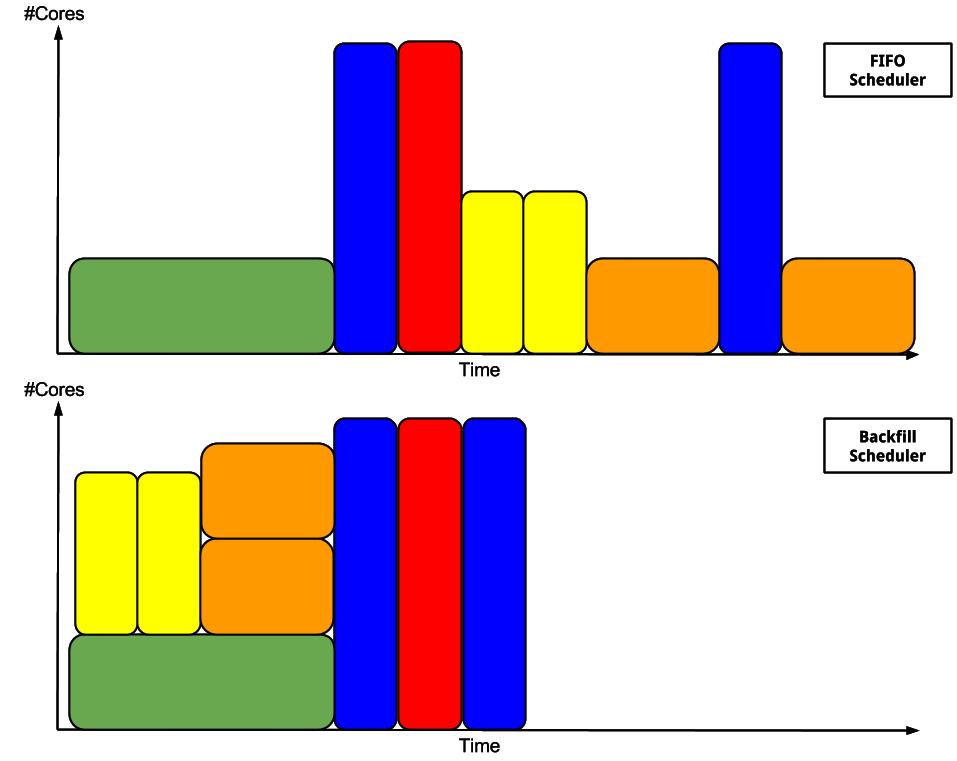
To benefit of the backfill allocation, you can specify a minimum and a maximum execution time when you submit a job. If the resources for the max execution time are not available, the scheduler will try to decrease the execution time until it reaches the minimum execution time. In the following example, we submit a job specifying that we want ideally a two days execution time with a one day minimum:
srun --tasks=128 --time-min=1-00:00:00 --time=2-00:00:00 --partition shared-cpu myjob
Attention
Warning : Be sure that you have some kind of checkpointing activated as your job will be terminated between the min and max execution time.
hpc/slurm.txt · Last modified: by Adrien Albert